Por primera vez, un estudio internacional liderado por el Instituto de Biología Evolutiva (IBE), un centro mixto del Consejo Superior de Investigaciones Científicas (CSIC) y la Universidad Pompeu Fabra (UPF), junto con la Universidad de São Paulo, ha descifrado el genoma de poblaciones indígenas americanas.
La investigación parte del Proyecto de Diversidad Genómica Indígena Americana (Indigenous American Genomic Diversity Project, IAGDP) y reúne la mayor base de datos genómica hasta la fecha. Sus resultados, publicados en Nature, arrojan luz sobre su historia y aportan nuevas claves sobre la salud y la evolución humana.
Los 128 genomas completos de alta cobertura secuenciados proceden de ocho países latinoamericanos —Argentina, Bolivia, Brasil, Colombia, Ecuador, México, Paraguay y Perú—, y representan 45 poblaciones y 28 familias lingüísticas.
A estos datos se añadieron genomas de alta calidad de bases de datos preexistentes, alcanzando un total de 199 individuos indígenas contemporáneos, pertenecientes a 53 poblaciones y 31 familias lingüísticas. Además, fueron incorporados datos de ADN antiguo para investigar aspectos más profundos de su historia y evolución.
“Hasta ahora, se habían caracterizado genéticamente apenas dos poblaciones indígenas amazónicas que, por la particularidad de su ambiente y su aislamiento, resultaban poco representativas”, comenta Marcos Araújo Castro e Silva, investigador en el IBE y primer autor del estudio.
La investigación identificó más de un millón de variantes genéticas no observadas previamente en otras poblaciones, lo que revela una diversidad genética única. “En conjunto, los hallazgos del estudio muestran que el origen de los pueblos indígenas americanos y la formación de su diversidad genética fueron procesos mucho más complejos de lo que pensábamos”, explica el investigador a este periódico.
El continente americano presenta una gran diversidad de ambientes y presiones ecológicas, desde las selvas del Amazonas hasta las grandes altitudes de los Andes, lo que ha favorecido la selección de distintas variantes genéticas y ha contribuido a la adaptación de las poblaciones humanas a estos entornos. Se identificaron señales genéticas de selección natural relacionadas con la respuesta inmunitaria, el metabolismo, el crecimiento y la fertilidad.
Nuevos hallazgos en la historia de los pueblos originarios americanos
Los investigadores encontraron evidencia de múltiples dispersiones humanas desde Norteamérica hacia Sudamérica. El desplazamiento de poblaciones asiáticas hacia América, a través de Beringia, representó la última gran migración humana continental. A excepción de algunas poblaciones, generalmente situadas en el Ártico, todos los indígenas americanos actuales descienden de una migración que ocurrió hace aproximadamente 15.000 años.
La primera expansión ocurrió inmediatamente después de la entrada en Norteamérica. Pero hace alrededor de 9.000 años, se produjo una segunda oleada migratoria que reemplazó, al menos en parte, a la primera. Por primera vez, el estudio identifica una tercera oleada migratoria. “Involucró a poblaciones antiguas emparentadas con los pueblos indígenas actuales de Mesoamérica. Estas poblaciones comenzaron a expandirse hacia Sudamérica hace al menos 1.300 años y tuvieron un impacto muy importante en la diversidad genética de las poblaciones indígenas de Sudamérica y el Caribe, tanto antiguas como contemporáneas”, detalla Marcos Araújo Castro e Silva.
La investigación también confirma el profundo efecto del “cuello de botella” provocado por la colonización europea. “La diversidad genética actual es solo una fracción de la original, pues la colonización diezmó a las poblaciones indígenas en un 90%. Aun así, observamos continuidad genética de más de 9.000 años en algunas regiones”, indica a Innovaspain Tábita Hünemeier, investigadora principal del IBE y líder del estudio. “Vimos que este impacto fue muy diferente según la región: algunas poblaciones conservaron una diversidad genética más alta, mientras que otras presentan todavía hoy niveles muy bajos de diversidad, lo que puede aumentar el riesgo de enfermedades genéticas y también el impacto de otras enfermedades”.
Desvelada la genética ancestral de los indígenas americanos
El estudio revela también que alrededor del 2% del genoma de algunos pueblos indígenas americanos muestra afinidad genética con poblaciones de Australasia, como las de Australia, Nueva Guinea y las Islas Andamán. Esta conexión, presente en individuos sudamericanos con más de diez mil años de antigüedad y en proporciones muy similares, sugiere la influencia de una población asiática antigua no muestreada, denominada Ypykuéra (ascendencia Y) que se mezcló con los ancestros de estas poblaciones.
“Esta señal sigue siendo un misterio, pero sugiere que los ancestros de los pueblos indígenas americanos no procedían de una única población homogénea, sino que se originaron a partir de la mezcla de al menos dos oleadas migratorias distintas provenientes de Asia, o de una población ancestral asiática ya muy diversa genéticamente. En este estudio inferimos que esa señal se ha mantenido hasta la actualidad. Esto sugiere que esta ancestría llegó al continente junto con los primeros americanos, o muy poco después de su entrada”, aclara la investigadora.
“Observamos que la frecuencia de esa ascendencia Ypykuéra es muy similar en las distintas poblaciones analizadas, lo que puede indicar cierta ventaja adaptativa en alguna de esas regiones genómicas”, explica David Comas, investigador principal del IBE y catedrático e investigador en el Departamento de Medicina y Ciencias de la Vida (MELIS) de la UPF, que ha colaborado en el estudio.
El trabajo también confirma que entre el 1% y el 3% del genoma procede de homínidos arcaicos, como los neandertales y los denisovanos, una proporción similar a la de otras regiones, aunque con un patrón distintivo. Además, estos homínidos aportaron variantes genéticas que resultaron importantes para la adaptación al continente americano, como evidencian los indicios de selección natural hallados en el genoma.
Innovación técnica y humana
El proyecto tiene una importante carga innovadora, pero Hünemeier destaca que el I+D más relevante del estudio ha sido el humano. “Este trabajo es el resultado de décadas de colaboración entre investigadores de América Latina y comunidades indígenas de todo el continente. Sin esas relaciones de confianza, construidas a largo plazo y basadas en la colaboración y el respeto, una investigación de esta magnitud simplemente no habría sido posible”.
Desde el punto de vista técnico, la investigación solo ha sido posible gracias a una combinación de avances que, hace apenas una o dos décadas, no existían o eran inaccesibles.
La secuenciación completa del genoma es ahora mucho más asequible. “Hace unos años, secuenciar genomas completos de centenares de personas habría sido prohibitivamente caro, especialmente para proyectos financiados mayoritariamente con recursos de países del Sur global, históricamente subrepresentados y marginados en la ciencia. Hoy podemos analizar el genoma completo de individuos de pueblos indígenas de todo el continente y estudiar no solo unas pocas variantes, sino también todo el espectro de su diversidad genética”, argumenta Hünemeier.
Además, los avances en bioinformática permitieron realizar análisis muy sofisticados. “Pudimos aislar los segmentos del genoma que proceden de ancestría indígena y compararlos con los de otras poblaciones actuales, así como con individuos antiguos. Esto nos ayuda a reconstruir con mucho mayor detalle la historia de las poblaciones americanas, sus movimientos, contactos y cambios demográficos a lo largo del tiempo”, añade.
La investigadora indica que otro elemento fundamental ha sido “el enorme desarrollo del ADN antiguo”. Y es que, durante los últimos años, se han generado y puesto a disposición pública grandes bases de datos con genomas de individuos que vivieron hace miles de años en distintas regiones de América y del mundo. “Gracias a ello, hoy podemos comparar directamente a las poblaciones indígenas contemporáneas con personas que vivieron en diferentes períodos y lugares del continente, algo que era impensable hace relativamente poco tiempo”.
“También han sido clave las nuevas metodologías estadísticas y computacionales desarrolladas en los últimos diez o veinte años”, apunta la experta.” La cantidad de datos genómicos que son generados actualmente es enorme, y necesitamos herramientas muy avanzadas para analizarlos. Estas metodologías nos permiten detectar señales muy sutiles de mezcla entre poblaciones, reconstruir parentescos antiguos, estimar cuándo ocurrieron determinados eventos demográficos e identificar señales de selección natural de variantes genéticas que antes pasaban desapercibidas.
El precio de ser ignorados
El equipo investigador identificó más de un millón de variantes genéticas de una sola base (en inglés single nucleotide variants o SNVs) que no están representadas en las bases de datos actuales.
Este punto es de especial relevancia en el conjunto de la investigación. Como explica Tábita Hünemeier, la investigación sobre la diversidad genómica humana tiene múltiples aplicaciones en biomedicina, evolución e historia. “Sin embargo, muchas poblaciones han estado históricamente infrarrepresentadas en el mapa genómico humano. Es el caso de las poblaciones indígenas americanas, cuya historia de adaptación y diversidad genética sigue siendo, en gran medida, desconocida”.
Asegura que los resultados obtenidos demuestran la necesidad de representar mejor a estas poblaciones en la genómica. “Conocer la diversidad genómica humana beneficia tanto a las comunidades indígenas como a la población global”. El equipo de Hünemeier describió en 2023 la resistencia genética al Chagas en poblaciones amazónicas y lideró el proyecto “DNA do Brasil”, publicado en 2025.
Entre los principales beneficios potenciales de este trabajo para la salud a los que alude la investigadora, está la posibilidad de mitigar un problema importante de la medicina genómica actual. “La enorme mayoría de los estudios genéticos se han hecho en personas de ascendencia europea, mientras que las poblaciones indígenas americanas han estado prácticamente ausentes. Como consecuencia, muchas pruebas genéticas, estimaciones de riesgo o incluso diagnósticos funcionan peor para personas con ancestría indígena o mestiza”.

Al generar uno de los catálogos más completos de variación genética indígena americana, “nuestro trabajo permite interpretar mucho mejor qué variantes son normales y frecuentes en estas poblaciones y cuáles podrían estar relacionadas con enfermedad”. Según la experta, esto puede ayudar, por ejemplo, a reducir falsos diagnósticos, evitar clasificar erróneamente variantes benignas como patológicas y mejorar el diagnóstico de enfermedades raras o hereditarias.
Por otro lado, detalla que estos datos pueden ser muy útiles para la medicina de precisión. “Muchas herramientas actuales para predecir el riesgo de enfermedades (por ejemplo, diabetes, enfermedades cardiovasculares o algunos tipos de cáncer) están basadas en datos europeos y, por tanto, son mucho menos precisas en personas latinoamericanas o con ancestría indígena. Incorporar esta diversidad genética permitirá desarrollar predicciones y tratamientos más ajustados a las poblaciones de América”.
El estudio también puede contribuir a identificar variantes genéticas con posibles implicaciones biomédicas que hasta ahora eran desconocidas. “Algunas de las variantes que encontramos podrían influir en la respuesta a medicamentos, en la susceptibilidad a determinadas enfermedades o en rasgos biológicos relevantes para la salud. Aunque todavía hará falta investigación adicional para entender exactamente su efecto, este trabajo crea una base imprescindible para futuros estudios médicos y farmacogenómicos”.
Adicionalmente, la comparación con ADN antiguo les permite entender cómo determinados rasgos biológicos o adaptaciones surgieron y se distribuyeron en las poblaciones americanas. “Puede ser relevante para estudiar, entre otras cosas, adaptaciones relacionadas con la dieta, el metabolismo, la respuesta inmunitaria o la vida en determinados ambientes”.
Como conclusión, Tábita Hünemeier opina que el beneficio más importante es que este trabajo contribuye a una medicina más equitativa. “Históricamente, las poblaciones indígenas y latinoamericanas han quedado excluidas de la investigación biomédica, lo que hace que muchas de las herramientas clínicas actuales resulten menos útiles para ellas. Incorporar esta diversidad no solo mejora el conocimiento científico, sino que ayuda a que los beneficios de la medicina genómica lleguen también a estas poblaciones”.
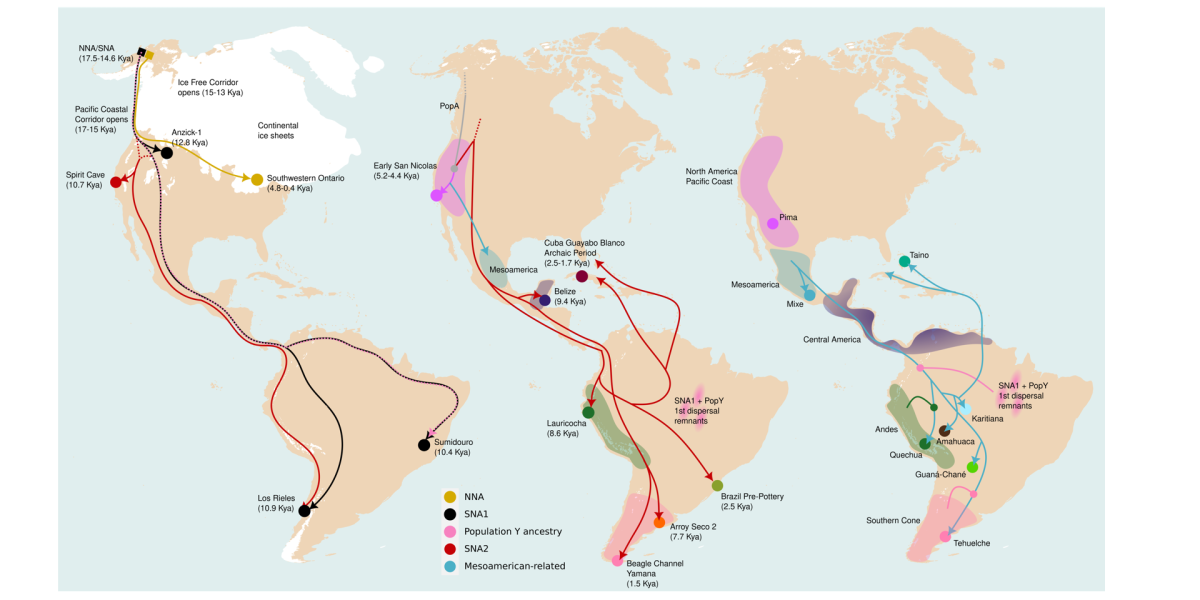
Nota: explicación de la imagen de cabecera.
De izquierda a derecha, cada panel representa una de estas dispersiones.
Los círculos indican la ubicación aproximada de individuos antiguos o poblaciones actuales, y las flechas señalan las rutas de dispersión.
El panel izquierdo muestra la primera dispersión (>9 mil años) y las primeras divisiones entre los ancestros de los indígenas americanos: Norteños (NNA) y Sureños (SNA), y dentro de estos últimos, dos ramas (SNA1 y SNA2). También incluye la contribución de la hipotética Población Y.
El panel central muestra la segunda dispersión (<9 mil años), asociada principalmente a SNA2, que reemplazó parcialmente a poblaciones previas, aunque algunos grupos con aporte de Población Y persistieron. Varias regiones muestran continuidad genética durante miles de años (zonas sombreadas).
El panel derecho muestra la tercera dispersión (<1,3 mil años), asociada a poblaciones mesoamericanas, que contribuyó ampliamente a Sudamérica y el Caribe. Estas poblaciones se mezclaron con grupos de la primera dispersión (incluyendo aporte de la Población Y) y, en los Andes y el Cono Sur, principalmente con poblaciones de la segunda dispersión, manteniendo una continuidad genética de hasta 9 mil años en estas regiones.



